Inhibiting SARS-CoV-2 Main Protease
The development of a new drug is a costly and lengthy process. Today as the world is facing a pandemic there is an urgent need to identify quickly drugs that stop virus proliferation. Drug repurposing offers an attractive alternative to this lengthy process by trying to identify if a drug known to be safe in humans could be used to treat new diseases.
While using such repositioned drugs individually might eventually not result in a significant clinical benefit, careful combination of drugs targeting several proteins crucial for virus replication and proliferation could be very effective, as was the case with HIV in the 1990s. The urgent question is which combination would be more effective?
Here we try to understand the structure of the SARS-CoV-2 protease active site, comparing it with existing structures of SARS-CoV protease complexed with micro-molar inhibitors, so that we can better understand the key interactions required to create a good inhibitor for SARS-CoV-2 protease.
We then conducted a virtual screening experiment using a library of FDA-approved drugs to see if some of these are predicted to bind to the protease. We examined how they are predicted to bind to the SARS-CoV-2 protease and could thus be used in a combination therapy.
SARS-CoV–2 Proteins
The SARS-CoV–2 genome from ill patients was quickly isolated and sequenced, providing the sequences of possible protein targets. These proteins share high sequence similarity with SARS-CoV proteins and initially research groups started building homology models. Now we are seeing more and more of these experimentally derived (X-ray and Cryo-EM) structures become available.
One of the best-characterized drug targets among coronaviruses is the main protease: Mpro, also called 3CL Protease.1 Along with the papain-like proteases, this enzyme is essential for processing the polyproteins that are translated from the viral RNA.2 It cleaves the amino acid backbone at 11 cleavage sites on the large polyprotein 1ab (Fig. 1).

Inhibiting the activity of this enzyme would block viral replication. Since no human proteases with a similar cleavage specificity are known, inhibitors to this target are much less likely to be toxic and cause side effects.
Structure-based Drug Design for Drug Repurposing: Main Protease
This main protease is a homodimer (Fig. 2) and each subunit contains a His41/Cys145 catalytic dyad.
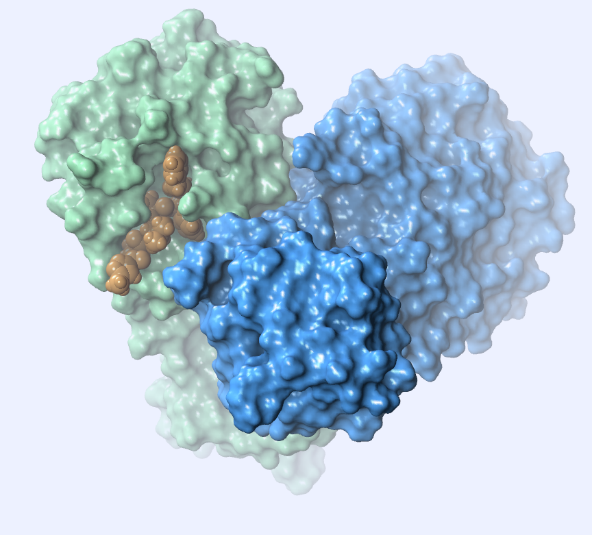
SARS-CoV-2 shares 96.1% identity and 99% similarity with SARS-CoV protease. (Fig. 3) There is only one amino acid difference in the active site: residue 46 in SARS-CoV-2 is a Serine instead of an Alanine in SARS-CoV.
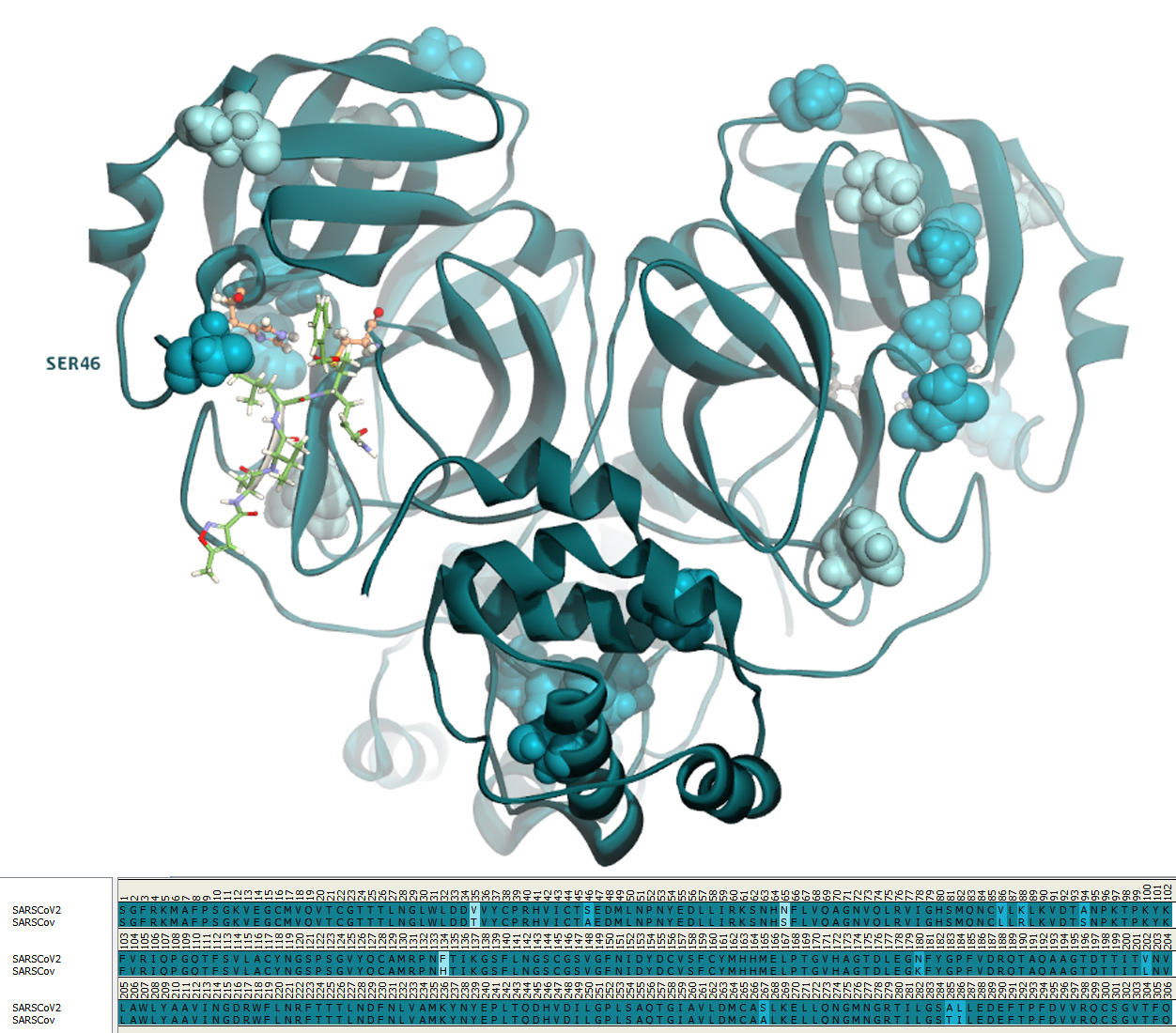
There many structures of SARS-CoV Mpro complexed with ligands available at the PDB and the ligands that have a micro molar binding affinity are all covalent ligands.
The active sites of the main protease are highly conserved amongst coronaviruses and are usually composed of four sites (S1′, S1, S2 and S4)3 (Fig. 4).
In the case of PDB structures 2ZU4 and 2GX4 for which ligands have respectively Ki values of 0.038 μM and 0.053 μM, the thiol of cysteine 145 in the S1′ site contacts inhibitors with a covalent bond. When compared with other inhibitors with lower affinity, this seems to be important for higher affinity.
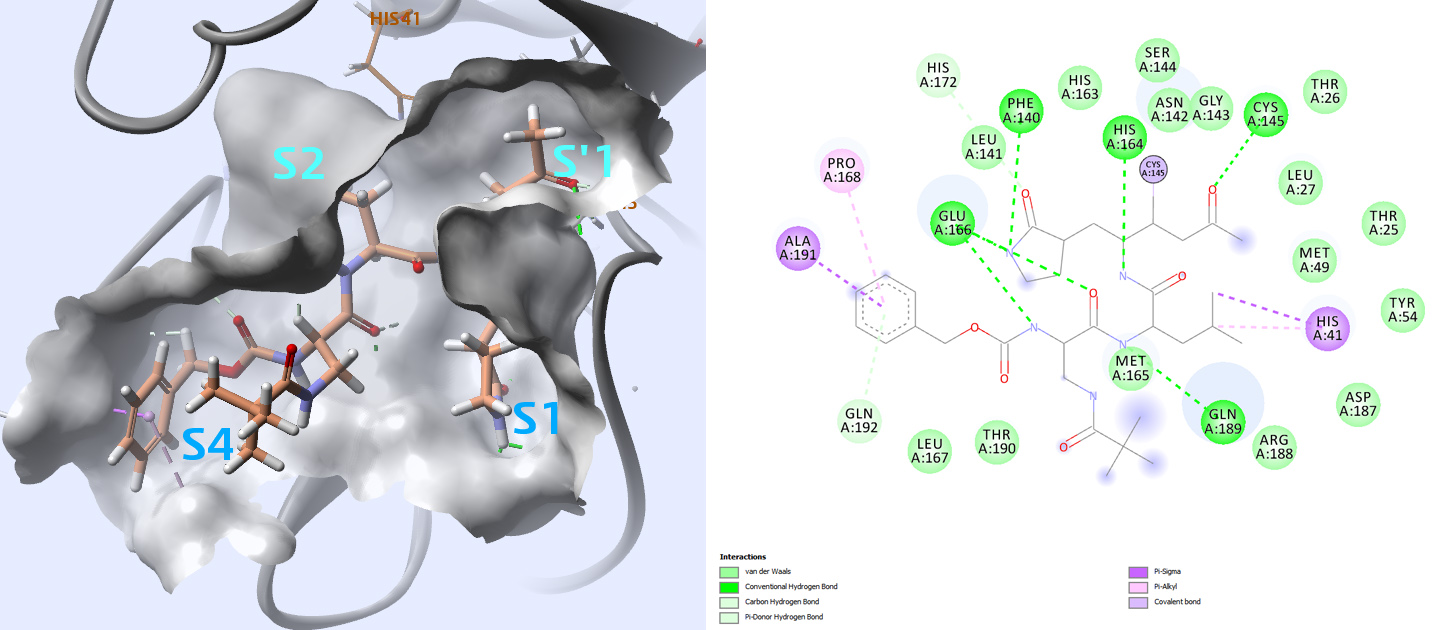
A. The inhibitor is displayed surrounded by the solvent accessible surface of the binding site. The four sub-sites S1,S’1,S2,S4 are labeled in blue.
B. In the 2D interaction plot, we can see that the ligand is covalently linked to the CYS145, and that it is interacting with residues: HIS 41, PHE140, GLU166, PRO168, GLN 189, ALA 191.
Careful examination of how all these ligands interact with this protease may provide information about key interactions to monitor when analyzing docking results.
Virtual Screening
Taking advantage of a high-resolution crystal structure of SARS-COV-2 protease dimer in complex with a covalently bonded peptide-like inhibitor N3 released in February (6LU7)4, we used this structure to conduct a virtual screening experiment. The inhibitor N3 that is co-crystalized binds in the substrate-binding pocket in an extended conformation. (Fig. 5)

We started with a library containing FDA-approved drugs with 2,684 compounds. We retained compounds with Molecular Weight lower than 800 kDa and performed docking with GOLD from the CCDC. Later, we calculated Binding Free Energies with CHARMM and GBMV implicit solvent. For each pose we first performed In Situ Ligand minimization with a sphere of 14 Å diameter for residue flexibility, and estimated Ligand Entropy when calculating the Binding Free Energy. Most of the compounds docked are known HCV and HIV protease drugs. (Fig. 6)
Many compounds make contacts in the 4 subsites: S1, S’1, S2, S4 and some with both HIS 41 and CYS 145, especially the HIV protease inhibitors.
The best scored compound is Troxerutin, which is a flavonoid.
- Flavonoids have been shown to inhibit some proteases5 and recently IC50 values were calculated from the dose-dependent inhibitory curves of herbacetin, rhoifolin and pectolinarin on SARS-CoV. The measured values were 33.17, 27.45 and 37.78 μM, respectively.
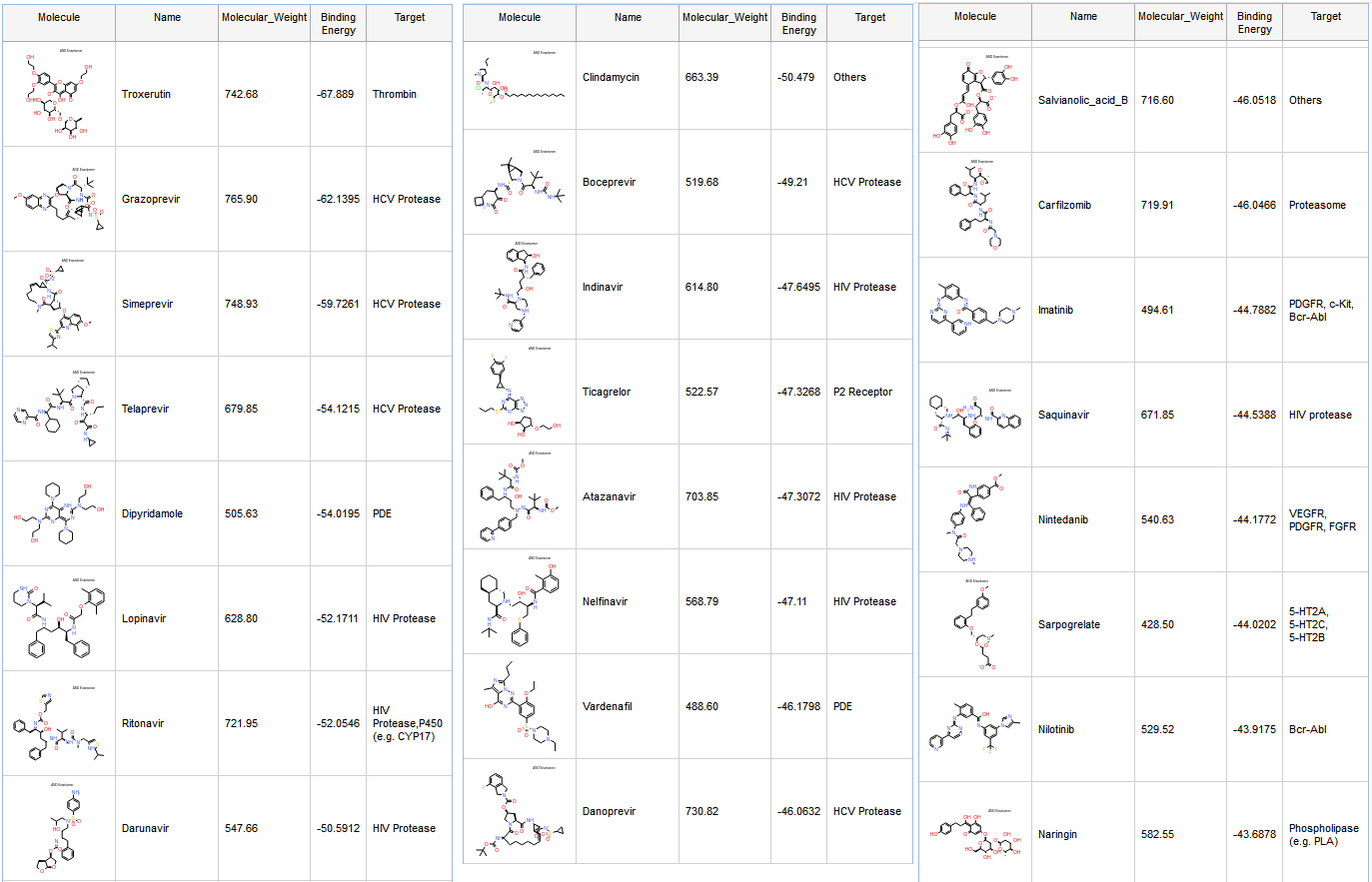
Dipyridamole (Fig. 7), which is the fifth ranked compound, has been mentioned recently in a preprint Therapeutic effects of dipyridamole on COVID-19 patients with coagulation dysfunction, and shown to suppress HCoV-19 replication at an EC50 of 100 nM in vitro.
Figure 7: 3D rendering and 2D interaction map of Dipyridamole in the main protease. Interactions with Catalytic residues are present, and with some of the residues mentioned earlier for SARS-CoV inhibitors.
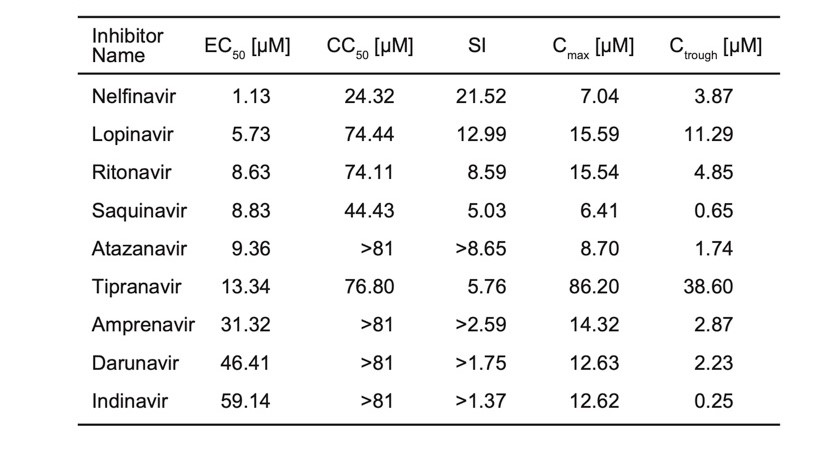
A recent preprint reported cellular assays of several HIV protease inhibitors: “Nelfinavir inhibits replication of severe acute respiratory syndrome coronavirus 2 in vitro.”
We are reporting the table from the preprint (Table 1). These are not IC50, but EC50, so it does not tell how well these molecules bind to the protease. However, this shows that all those compounds are inhibiting replication of SARS-CoV-2 and some are more active than others.
Ritonavir and Lopinavir are interacting with many residues previously seen in other compounds targeting SARS-CoV protease (Fig. 8 and 9). Although several clinical trials are ongoing with a combination of those two molecules, there are unfortunately no IC50 data available for Ritonavir or Lopinavir on SARS-CoV-2.
Figure 8: Best scored pose for Ritonavir: and 2D interaction diagram showing that Ritonavir is interacting with the two catalytic residues: CYS145 and HIS41, but also GLU166, PRO168 and GLN189 as seen with other known inhibitors of SARS-CoV.
Figure 9: Best scored pose for Lopinavir: and 2D interaction diagram showing that Lopinavir is interacting with the two catalytic residues: CYS145 and HIS41, but also GLU166, PRO168 and GLN189 as seen with other known inhibitors of SARS-CoV.
Nelfinavir (Fig. 10) has a high predicted binding affinity in our calculations and has also been characterized against COVID19 in a recent preprint : “Nelfinavir is active against SARS-CoV-2 in Vero E6 cells.“

A second preprint shows that Atazanavir is active on SARS-CoV-2 infected cells: “Atazanavir inhibits SARS-CoV-2 replication and pro-inflammatory cytokine production.”
A PDB structure 6W63 that contains a new inhibitor for SARS-CoV-2 was published after we started this work (Fig. 11). There is no published information about the binding affinity of this compound to the SARS-CoV-2 Mpro. The side chains in the binding site are almost in the same orientation as for 6LU7. We docked our compounds and rescored them with MMGBSA approaches as previously and found very similar results.
Figure 11: PDB structure 6W63: SARS-CoV-2 main protease in complex with inhibitor X77.
Perspectives
The binding site of SARS-CoV-2 protease is very large, containing four sub-sites, and can accommodate many different ligands with a moderate binding affinity. As explained here, virtual screening is a useful tool to identify possible inhibitors and was able to give us some structural explanation as to how possible inhibitors might be interacting with the protease.
However, all these hypotheses require confirmation by experimental evidence, like measurement of IC50, to build models that are more robust.
Here we have not performed covalent docking, but as the most active compounds against SARS-CoV protease were covalent, this might be a requirement of strong inhibitors. Covalent inhibitors also display advantages compared to reversible inhibitors such as having strong target affinity and prolonged acting lives in patients.
For a drug to be active in a patient, we need to consider other pharmacological aspects such as pharmacokinetics. It will be necessary to evaluate the ability of these drugs to achieve target plasma and lung concentrations following approved dosing in humans.
It was not within the scope of this work to comment on other structure-based design research, as this would not be timely for the current epidemic. Our purpose was to assess readily usable therapeutics for potential candidate drugs against COVID19.
We would like to thank the Cambridge Crystallographic Data Centre (CCDC) for permission to use GOLD for the virtual screening in this work. GOLD has proven success in virtual screening, lead optimization, and identifying the correct binding mode of active molecules. An interface to GOLD is available within Discovery Studio.
References
- K. Anand, J. Ziebuhr, P. Wadhwani, J. R. Mesters, R. Hilgenfeld. Coronavirus main proteinase (3CLpro) structure: Basis for design of anti-SARS drugs. Science. 2003, 300(5626):1763-7.
- R. Hilgenfeld. From SARS to MERS: Crystallographic studies on coronaviral proteases enable antiviral drug design. FEBS J. 2014, 281(18):4085-96.
- H. Yang et al. Design of wide-spectrum inhibitors targeting coronavirus main proteases. PLoS Biol. 2005, 3(10):e324.
- Jin, Z. et al. Structure of Mpro from COVID-19 virus and discovery of its inhibitors. Nature. 2020, doi: 10.1038/s41586-020-2223-y.
- Jo, S. et al. Inhibition of SARS-CoV 3CL protease by flavonoids J Enzyme Inhib Med Chem 2020 Dec;35(1):145-151. doi: 10.1080/14756366.2019.1690480